|
|
本人已委托维权骑士(http://www.rightknights.com)进行原创维权。
思想高屋建瓴,知识原创共享。
转载请引用链接。
<hr/>在之前的富集分析系列文章(糖糖家的老张:最全的GO, KEGG, GSEA分析教程(R),你要的高端可视化都在这啦![包含富集圈图])中,向大家介绍了GO、KEGG富集分析中各种天花乱坠的可视化来让你的文章或汇报更高端,但无论是GO(基因本体)数据库还是KEGG(京都基因和基因组百科全书)数据库,都只能在官方支持的物种(此处暂称为模式生物,实际是充分但不必要的关系)中进行分析与可视化。
这就带来了一个问题:如果科学家研究的物种比较小众,或者是一类变异的亚群,如何针对这种小众或非模式生物进行GO与KEGG富集分析呢?我咨询了两家小有名气的生物科技公司如何解决这个问题,公司表示无法提供官方数据库以外的个性化服务。
事实上,我们可以通过自己建立该物种的GO或KEGG的数据库来解决。
<hr/>首先我们先来看看GO和KEGG数据库官方支持的物种有哪些?
GO数据库支持19个物种的分析,实在是少得可怜,研究微生物的课题组那么多,提供生信服务的公司也不少,我问了一圈没想到大家的回答基本都是GO分析不支持我们就不做GO了。
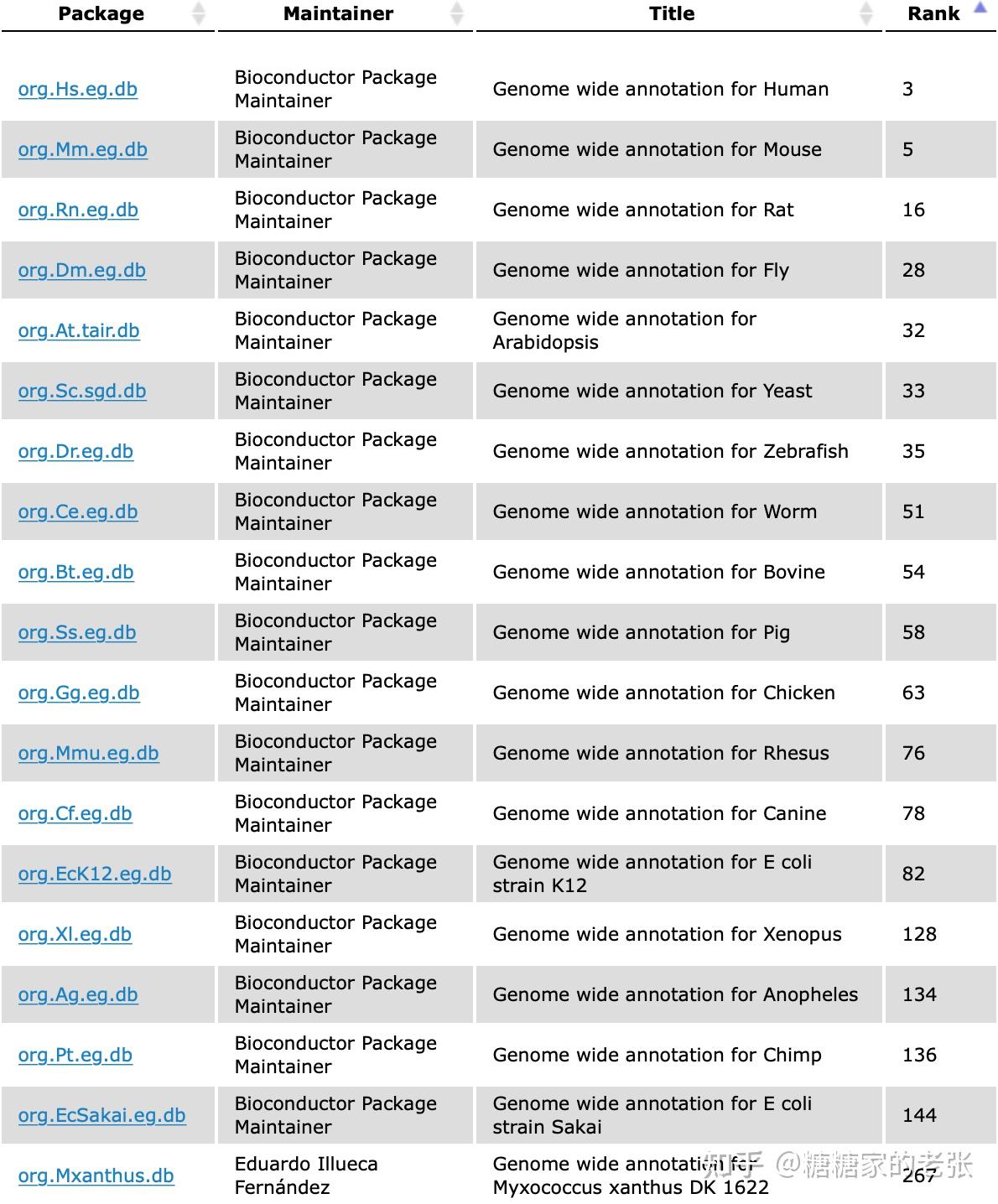
KEGG数据库支持的物种倒是很丰富,但是也难以做到适用任意种群的生物。
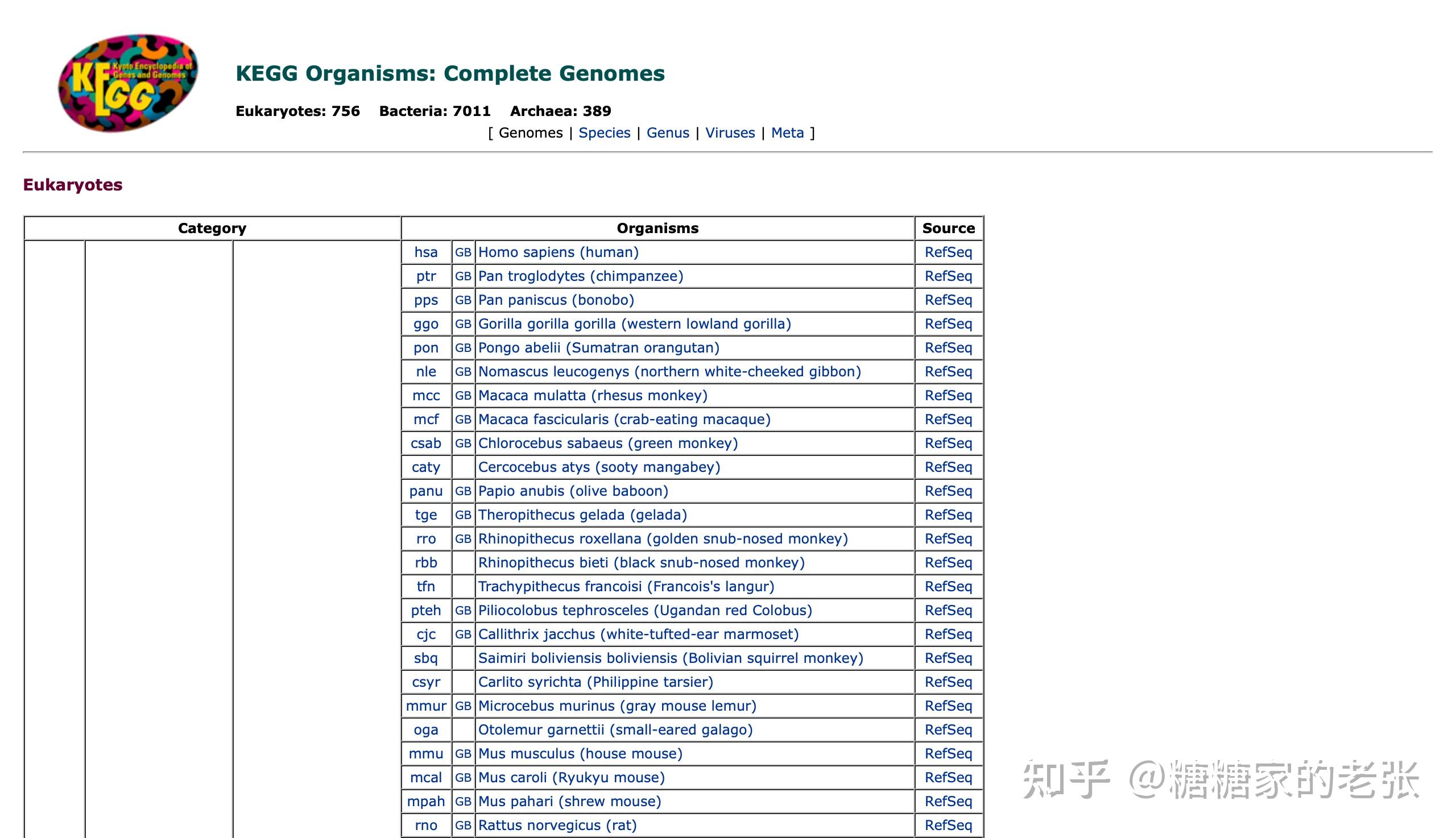
综上所述,GO和KEGG富集分析的使用之广而官方支持的物种之少都表明针对小众或非模式生物的自建库分析是研究和市场都亟待解决和普及的重要内容。接下来把思路和代码公开出来供大家参考。
<hr/>一.准备工作
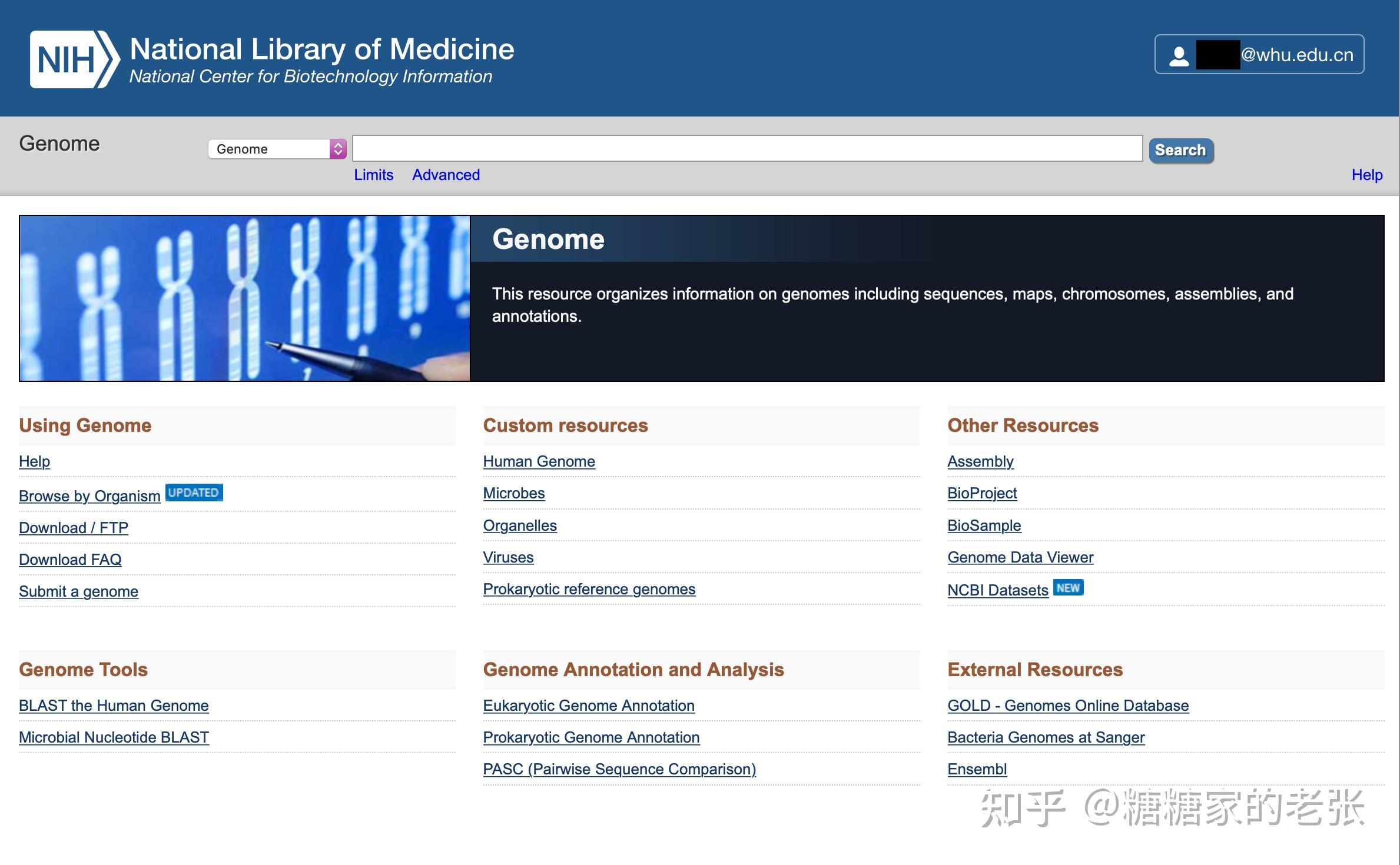
1.蛋白质组序列
从 https://www.ncbi.nlm.nih.gov/genome/ 可以检索和下载到所需物种的基因组及蛋白质组序列,当然,也可以通过其他方式自行准备。
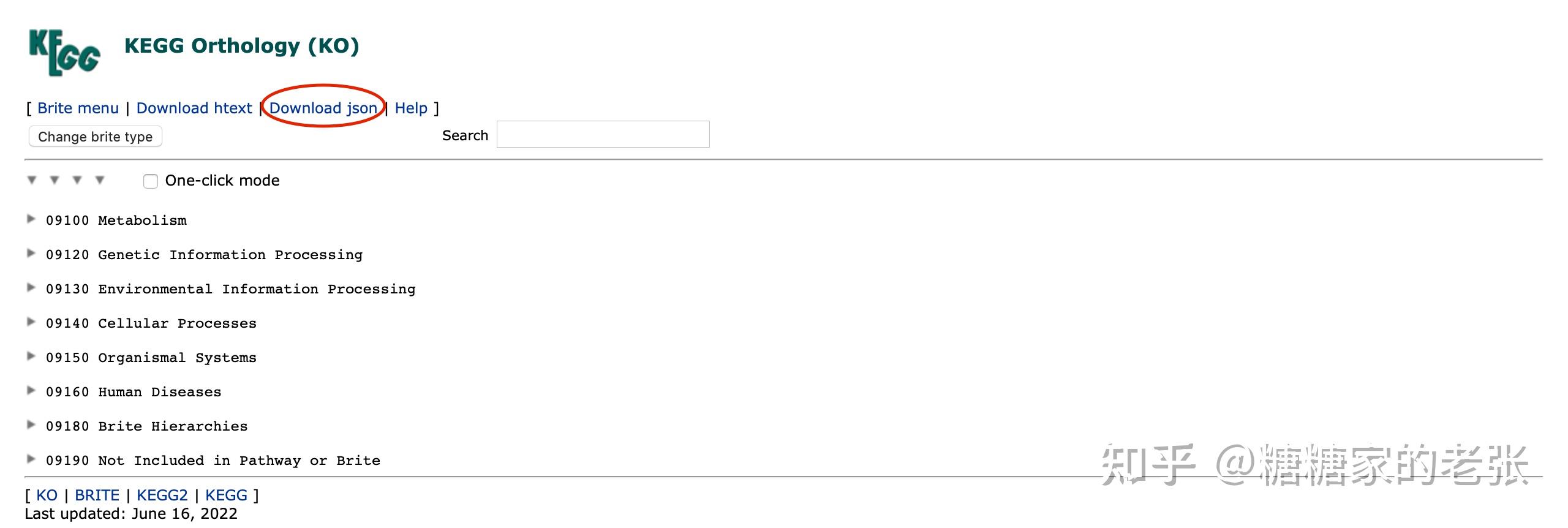
2.KEGG数据库官方的基因注释信息文件
从 可以下载到KEGG数据库对所有基因的注释信息json格式文件,关于为何要准备KEGG的注释信息这要从KEGG分析普及起了,可以去看我之前的文章糖糖家的老张:最全的GO, KEGG, GSEA分析教程(R),你要的高端可视化都在这啦![包含富集圈图]。
3.编程准备
R语言、R studio或其他编译器、以及如下R包的安装,具体如何安装可以参照我以前的教程。
library(openxlsx)
library(clusterProfiler)
library(dplyr)
library(stringr)
library(AnnotationForge)
library(jsonlite)
library(purrr)
library(RCurl)<hr/>二.自建GO数据库
1.蛋白质组注释
在egg-mapper网站进行蛋白质组的注释,直接提交蛋白质组序列文件即可,具体原理可以参照该工具的文献,大致就是把蛋白组中的蛋白和已收录进ncbi和uniport的蛋白进行相似性比对和聚类从而给蛋白注释功能。
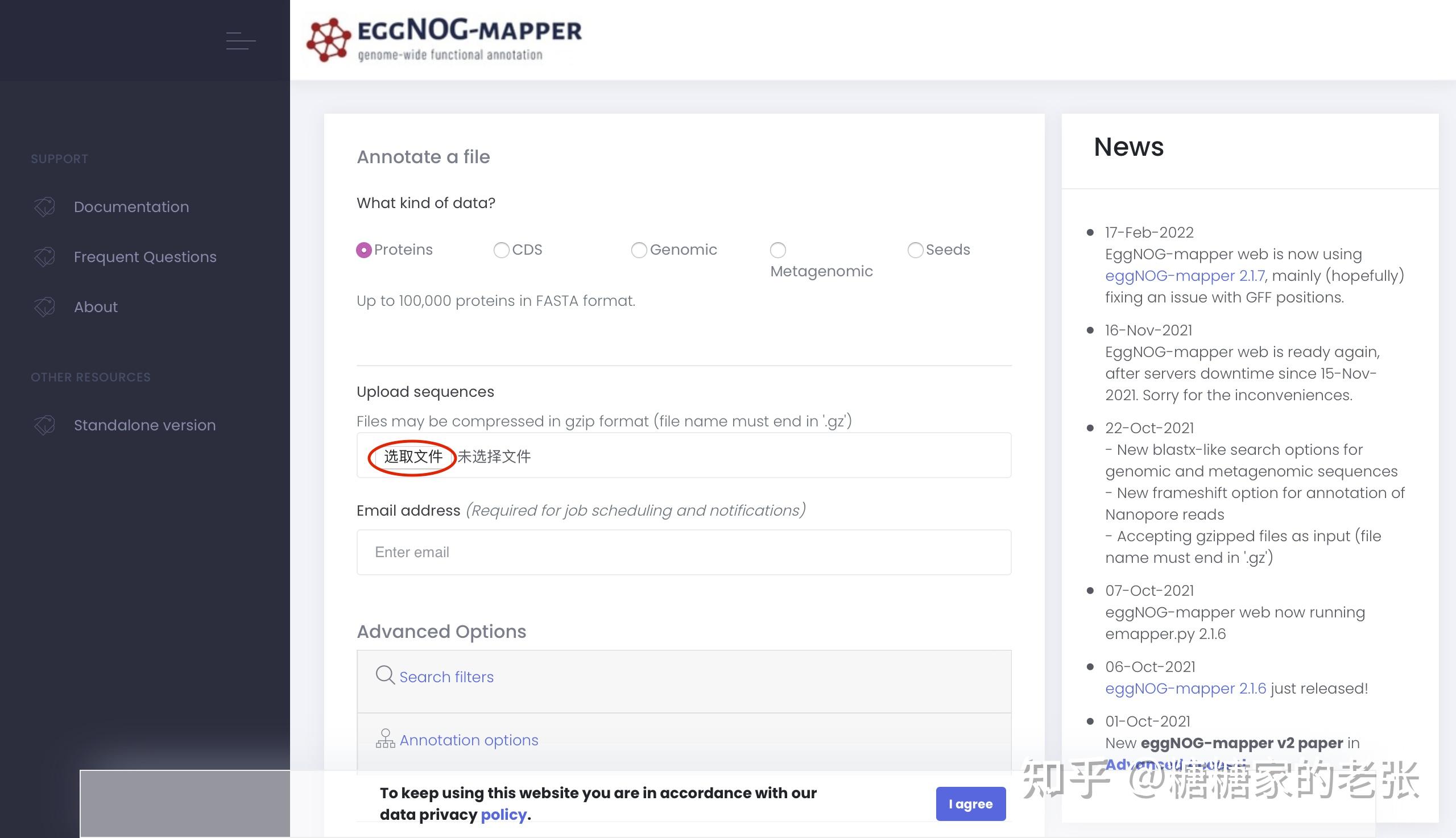
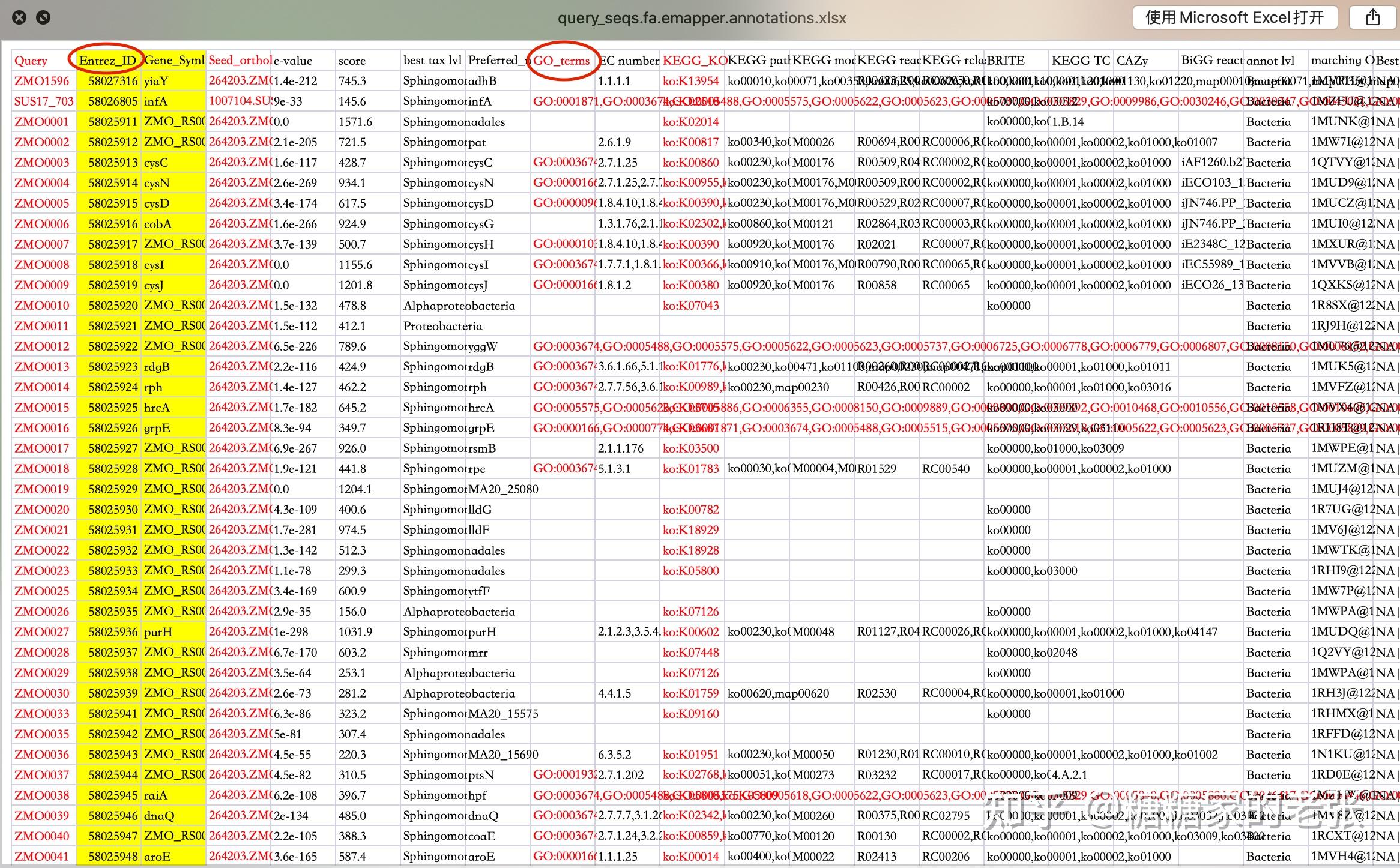
完成注释后下载xlsx格式的注释文件,在EXCEL中打开进行以下修改:
- 删除注释文件开头和结尾由#开头的几行
- 将列名GO terms改为GO_terms,这是为了方便R读取
- 将Query这列RefSeq格式序列号在 https://biodbnet-abcc.ncifcrf.gov/db/db2db.php 转换为ENTREZ ID即GENE ID并将该列加入表格(也可以选择别的基因ID转换工具进行ID转换)
最终得到的表格如下:
2.代码建库
R运行以下代码读取刚刚修改完的蛋白组注释文件:
library(openxlsx)#读取.xlsx文件
library(clusterProfiler)
library(dplyr)
library(stringr)
library(AnnotationForge)
options(stringsAsFactors = F)
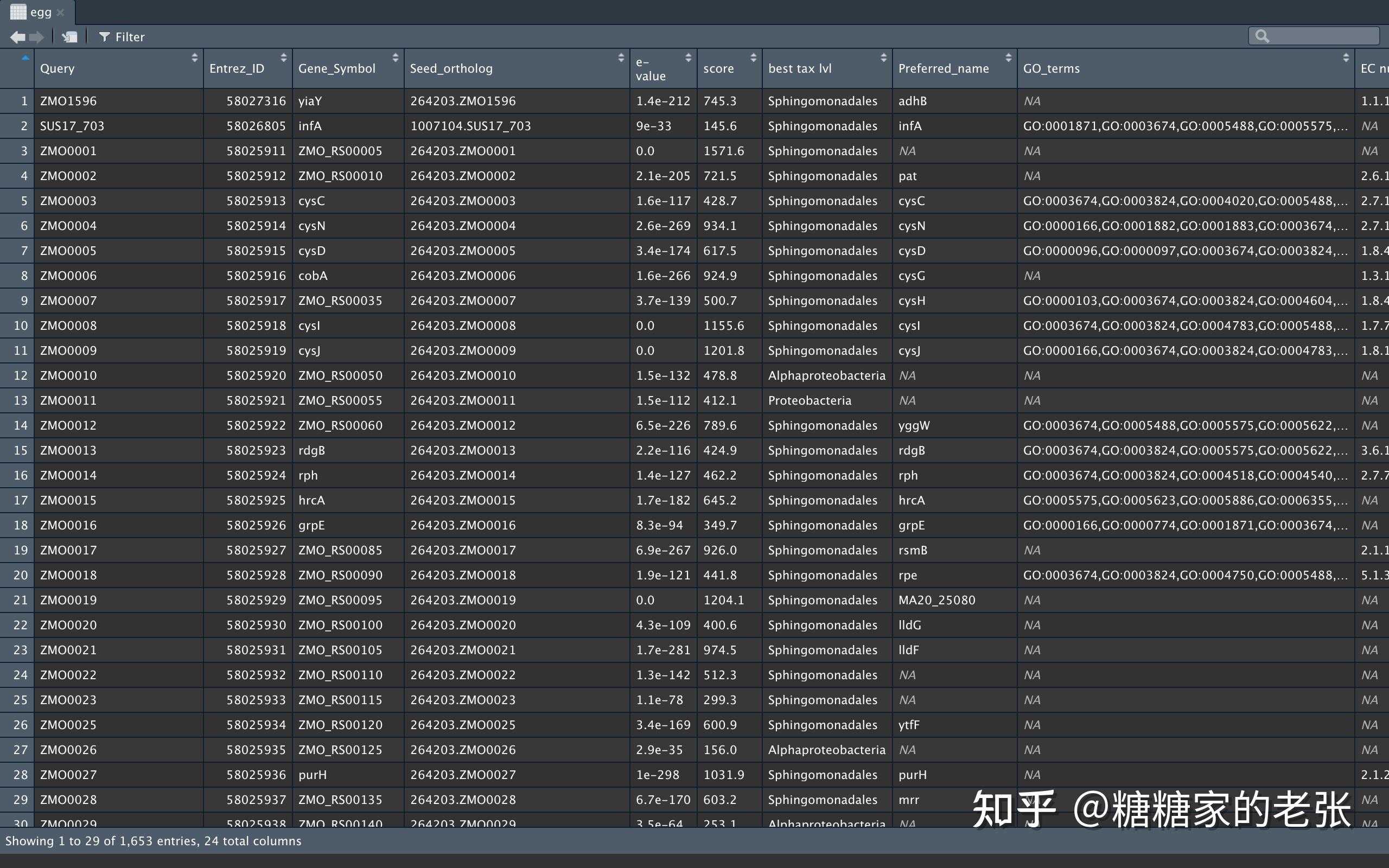
#########读取蛋白组注释文件#########
egg_f <- &#34;/Users/ZYP/SCIENCE/SCRIPTS/R/orgdb/eggNOG_annotation/ZM4/query_seqs.fa.emapper.annotations.xlsx&#34;
egg <- read.xlsx(egg_f, sep = &#34;\t&#34;)
egg[egg==&#34;&#34;]<-NA#将空行变成NA以便去除注释文件格式如下:
运行以下代码提取蛋白名称(Query)与egg注释信息(seed_ortholog)并建立关联:
#########提取蛋白名称(Query)与egg注释信息(seed_ortholog)#########
gene_info <- egg %>% dplyr::select(GID = Query, EGGannot = Seed_ortholog, SYMBOL=Gene_Symbol, ENTREZID=Entrez_ID) %>% na.omit()得到的数据格式如下:

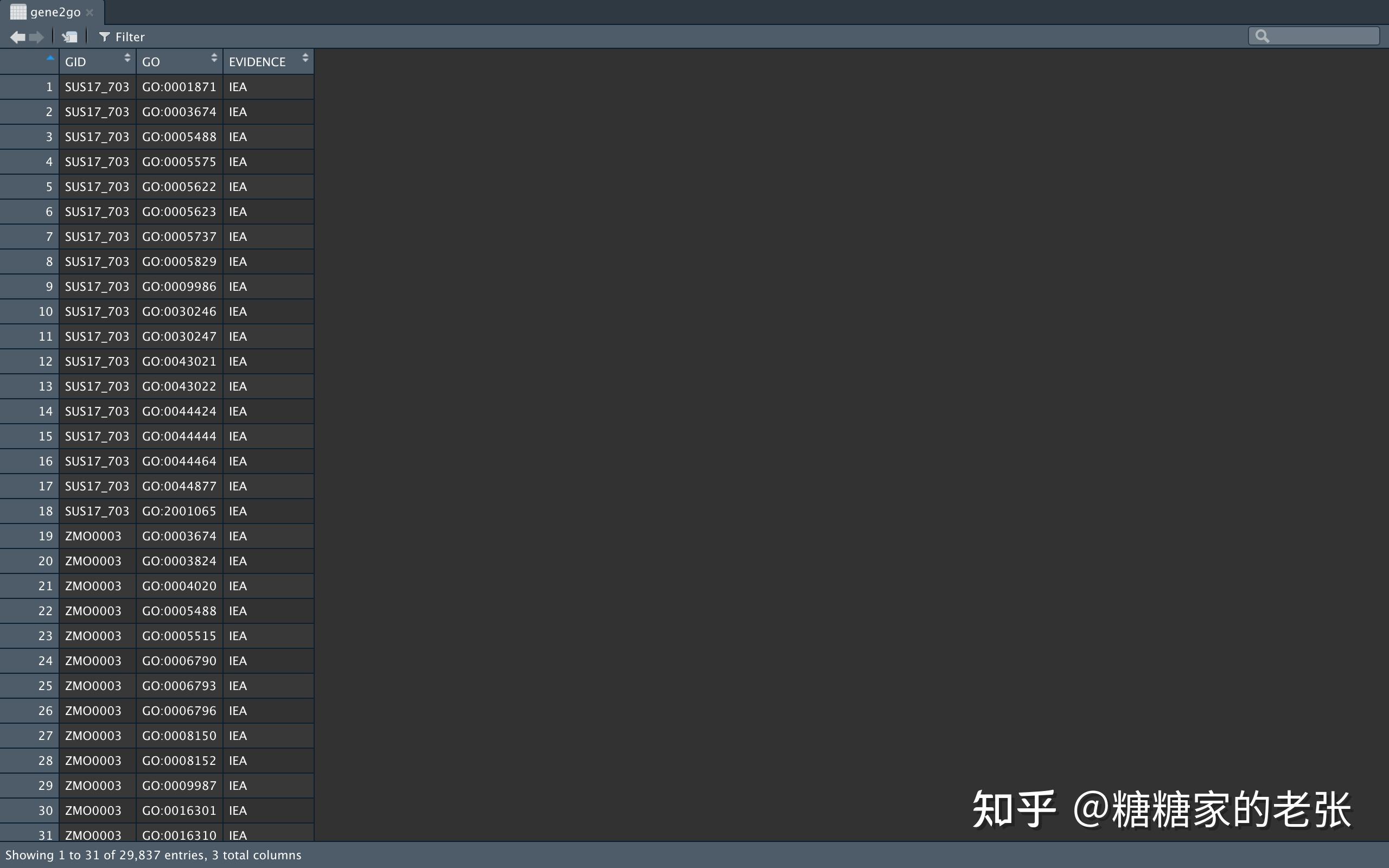
运行以下代码提取蛋白名称(Query)与GO注释信息(GO_terms)并建立关联:
#########提取蛋白名称(Query)与GO注释信息(GO_terms)#########
gterms <- egg %>% dplyr::select(GID = Query,GO_terms = GO_terms) %>% na.omit()
gene2go <- data.frame(GID = character(),#由于此时存在一蛋白对应多个GO,因此将其拆成一对一的多列储存进新的dataframe中
GO = character(),
EVIDENCE = character())
for (row in 1:nrow(gterms)) {
gene_terms <- str_split(gterms[row,&#34;GO_terms&#34;], &#34;,&#34;, simplify = FALSE)[[1]]
gene_id <- gterms[row, &#34;GID&#34;][[1]]
tmp <- data.frame(GID = rep(gene_id, length(gene_terms)),
GO = gene_terms,
EVIDENCE = rep(&#34;IEA&#34;, length(gene_terms)))
gene2go <- rbind(gene2go, tmp)} 得到的数据格式如下:
运行以下代码建立GO格式的数据库并保存用于后续分析,其中的数据库信息如物种名、种群名、ID、维护信息等请大家根据#号后的说明自行修改:
#########建库保存#########
genus = &#34;Zymomonas mobilis&#34;
species = &#34;ZM4&#34;
makeOrgPackage(gene_info=gene_info,go=gene2go,
version=&#34;0.1&#34;,#指定该GO富集注释数据库的版本号
maintainer = &#39;Y.-P.Z. <444764089@qq.com>&#39;,#指定该数据库维护者信息
author = &#39;Y.-P.Z. <444764089@qq.com>&#39;,#指定该数据库作者信息
outputDir = &#34;/Users/ZYP/SCIENCE/SCRIPTS/R/orgdb/database/ZM4/&#34;,#数据库的保存路径
tax_id = &#34;264203&#34;,#查询物种的Taxonomy https://www.ncbi.nlm.nih.gov/taxonomy
genus = genus,
species = species,
goTable=&#34;go&#34;)
zymo_ZM4.orgdb <- str_c(&#34;org.&#34;, str_to_upper(str_sub(genus, 1, 1)) , species, &#34;.eg.db&#34;, sep = &#34;&#34;)#将数据库命名为genus首字母+species全称
<hr/>三.自建KEGG数据库
KEGG自建库的过程与GO自建库的思路一致,只是代码读取的信息略有不同,此处就将各过程和注释组合为一个完整工作流进行展示:
library(jsonlite)
library(purrr)
library(RCurl)
options(stringsAsFactors = F)
#########读取蛋白组注释文件与通路注释文件#########
egg_f <- &#34;/Users/ZYP/SCIENCE/SCRIPTS/R/orgdb/eggNOG_annotation/ZM4/query_seqs.fa.emapper.annotations.xlsx&#34;
egg <- read.xlsx(egg_f, sep = &#34;\t&#34;)
egg[egg==&#34;&#34;]<-NA#将空行变成NA以便去除
KEGG_info=&#39;/Users/ZYP/SCIENCE/SCRIPTS/R/orgdb/KEGG_annotation/ko00001.json&#39;#from https://www.genome.jp/kegg-bin/get_htext?ko00001
#########提取蛋白名称(Query)与KEGG注释信息(KEGG_KO)#########
gene2ko <- egg %>% dplyr::select(GID = Query, Ko = KEGG_KO) %>% na.omit()
#########提取通路(Pathway)与通路名称(Name)信息#########
pathway2name <- tibble(Pathway = character(), Name = character())
ko2pathway <- tibble(Ko = character(), Pathway = character())
kegg <- fromJSON(KEGG_info)
for (a in seq_along(kegg[[&#34;children&#34;]][[&#34;children&#34;]])) {
A <- kegg[[&#34;children&#34;]][[&#34;name&#34;]][[a]]
for (b in seq_along(kegg[[&#34;children&#34;]][[&#34;children&#34;]][[a]][[&#34;children&#34;]])) {
B <- kegg[[&#34;children&#34;]][[&#34;children&#34;]][[a]][[&#34;name&#34;]][]
for (c in seq_along(kegg[[&#34;children&#34;]][[&#34;children&#34;]][[a]][[&#34;children&#34;]][][[&#34;children&#34;]])) {
pathway_info <- kegg[[&#34;children&#34;]][[&#34;children&#34;]][[a]][[&#34;children&#34;]][][[&#34;name&#34;]][[c]]
pathway_id <- str_match(pathway_info, &#34;ko[0-9]{5}&#34;)[1]
pathway_name <- str_replace(pathway_info, &#34; \\[PATH:ko[0-9]{5}\\]&#34;, &#34;&#34;) %>% str_replace(&#34;[0-9]{5} &#34;, &#34;&#34;)
pathway2name <- rbind(pathway2name, tibble(Pathway = pathway_id, Name = pathway_name))
kos_info <- kegg[[&#34;children&#34;]][[&#34;children&#34;]][[a]][[&#34;children&#34;]][][[&#34;children&#34;]][[c]][[&#34;name&#34;]]
kos <- str_match(kos_info, &#34;K[0-9]*&#34;)[,1]
ko2pathway <- rbind(ko2pathway, tibble(Ko = kos, Pathway = rep(pathway_id, length(kos))))
}
}
}
#########组建通路(Pathway)与蛋白名称(Query)信息#########
ko2gene <- tibble(Ko=character(),GID=character())#由于此时存在一蛋白对应多个KO,因此将其拆成一对一的多列储存进新的dataframe中
for (Query in gene2ko$GID){
ko_list <- strsplit(gene2ko$Ko[which(gene2ko[,1]==Query)],split = &#39;,&#39;)
for (ko in ko_list){
if (length(which(ko2gene[,1]==ko))==0){
tmp <- data.frame(Ko=ko,GID=Query)
ko2gene <- rbind(ko2gene,tmp)
}
else{
old_Query <- ko2gene$GID[which(ko2gene[,1]==ko)]
ko2gene$GID[which(ko2gene[,1]==ko)] <- paste(old_Query,Query,sep = &#39;,&#39;)
}
}
}
pathway2gene <- tibble(Pathway = character(), GID = character())
for (ko in ko2pathway$Ko){
pathway_list <- ko2pathway$Pathway[which(ko2pathway[,1]==ko)]
for (pathway in pathway_list){
if (paste(&#39;ko:&#39;,ko,sep=&#39;&#39;) %in% ko2gene$Ko){
ko <- paste(&#39;ko:&#39;,ko,sep=&#39;&#39;)
if (length(which(pathway2gene[,1]==pathway))==0 ){
ko2gene$GID[which(ko2gene[,1]==ko)]
tmp <- data.frame(pathway=pathway,GID=ko2gene$GID[which(ko2gene[,1]==ko)])
pathway2gene <- rbind(pathway2gene,tmp)
}
else{
old_Query <- pathway2gene$GID[which(pathway2gene[,1]==pathway)]
Query <- ko2gene$GID[which(ko2gene[,1]==ko)]
pathway2gene$GID[which(pathway2gene[,1]==pathway)] <- paste(old_Query,Query,sep=&#39;,&#39;)
}
}
}
}
#########建库保存#########
KEGG_database=&#39;/Users/ZYP/SCIENCE/SCRIPTS/R/orgdb/database/ZM4/KEGGdatabase_ZM4.Rdata&#39;
save(pathway2gene, pathway2name, file = KEGG_database)
<hr/>那么最后可以在保存路径下找到自己建立的该物种的GO和KEGG数据库如下:

<hr/>四.自建库富集分析
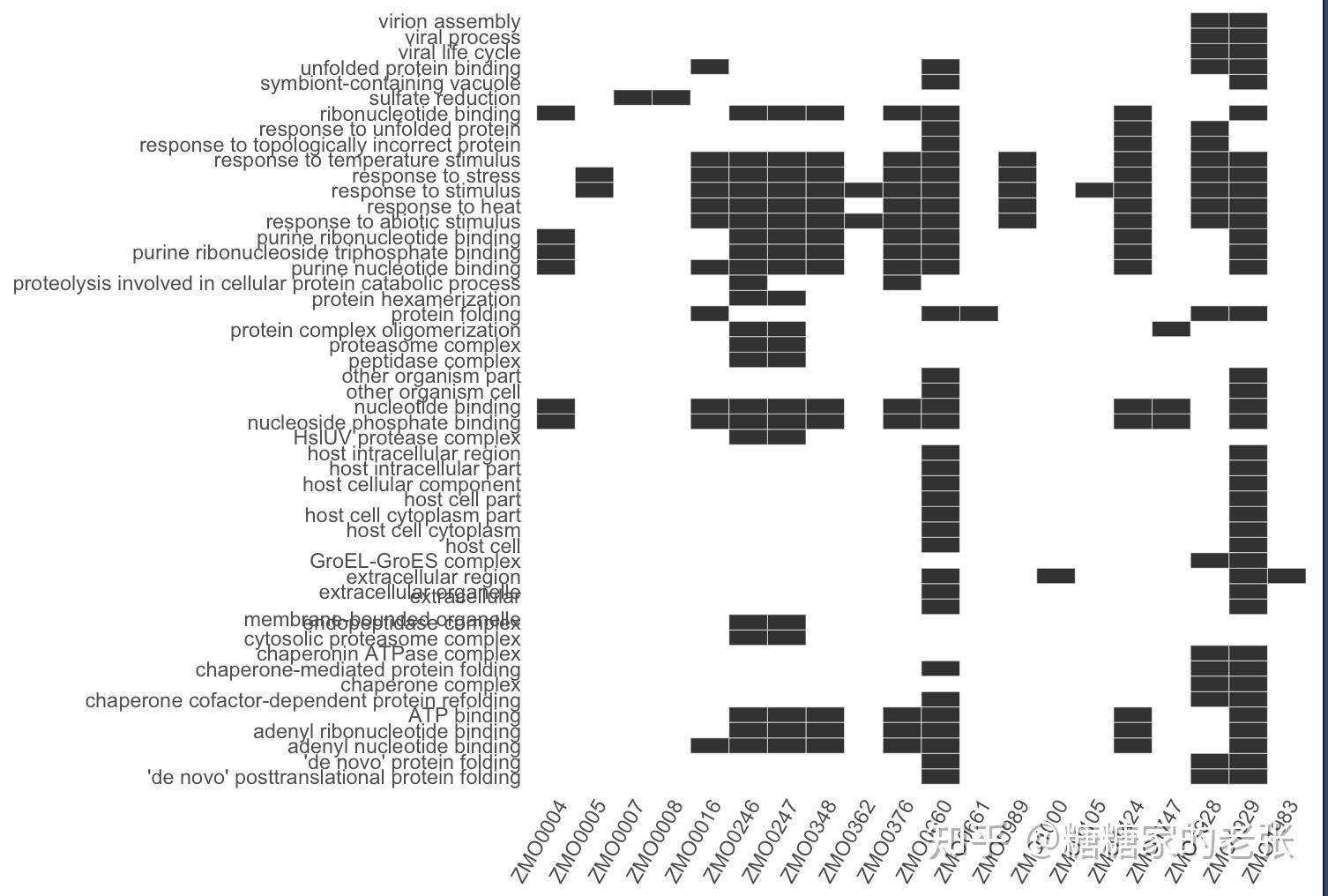
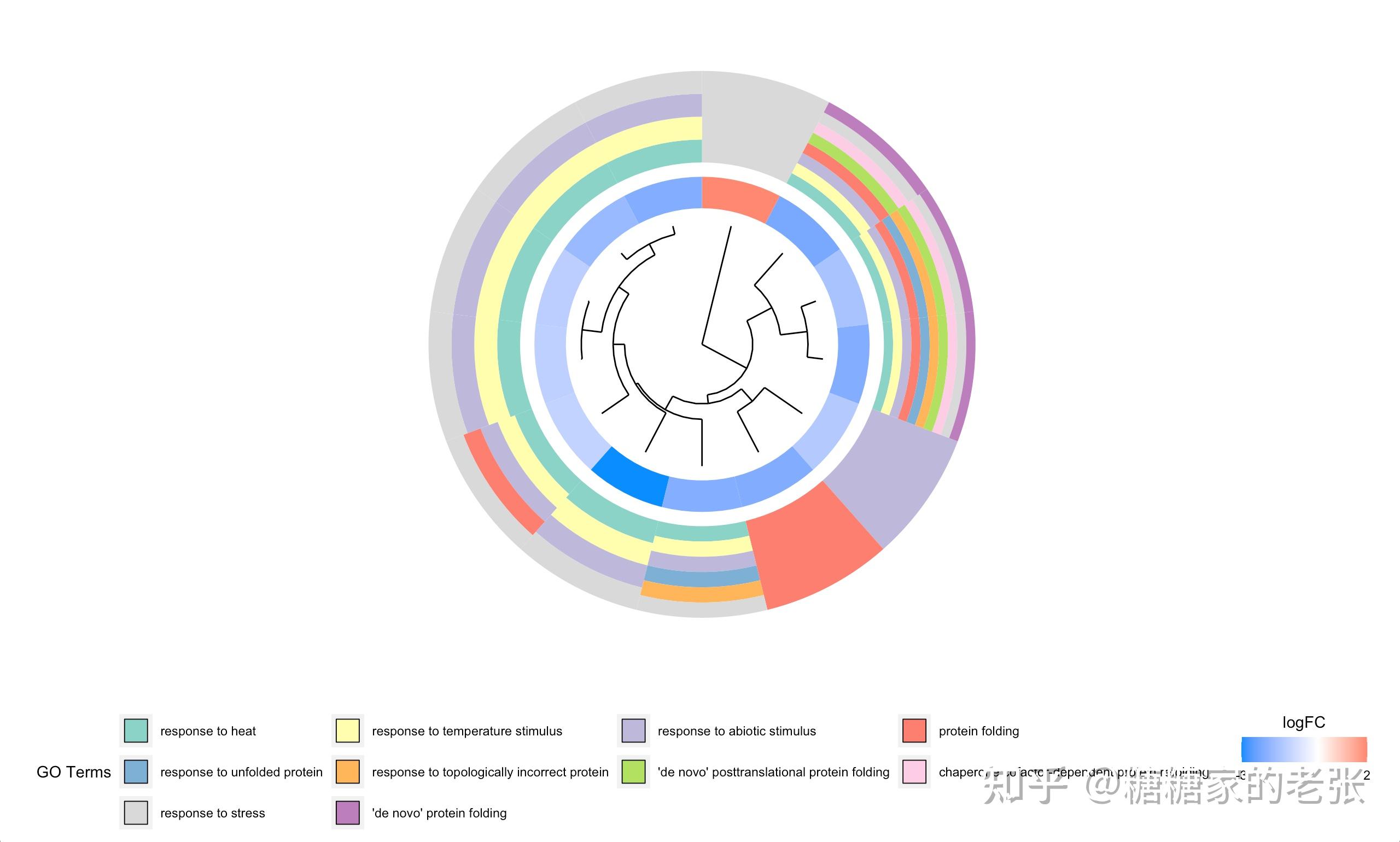
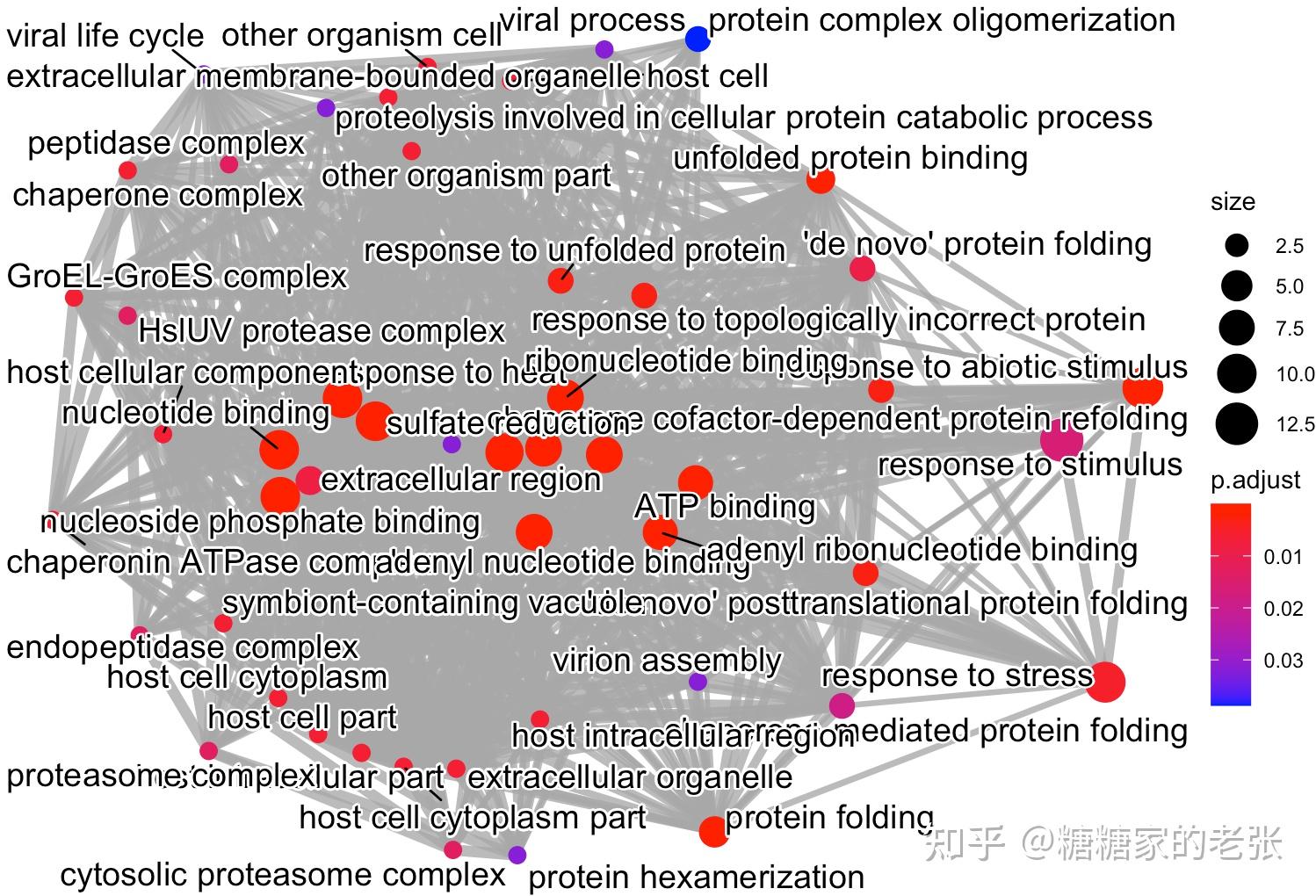
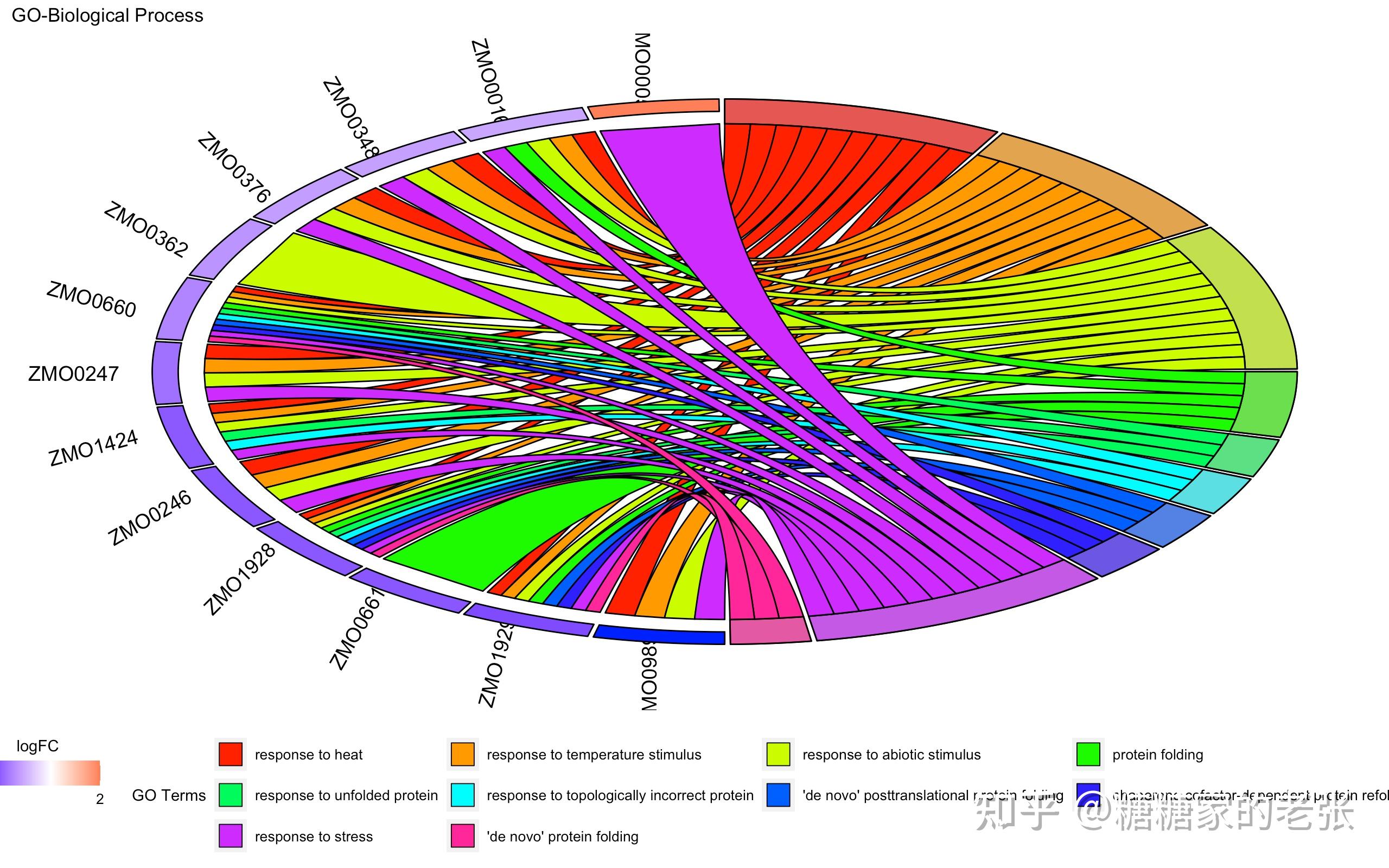
接下来大家就可以使用建立的数据库进行小众或非模式生物的GO和KEGG分析了,详细的GO和KEGG分析教程可以参照我之前的文章 糖糖家的老张:最全的GO, KEGG, GSEA分析教程(R),你要的高端可视化都在这啦![包含富集圈图] ,这里放出代码和几张自建库富集的示例结果图片:
library(openxlsx)#读取.xlsx文件
library(ggplot2)#柱状图和点状图
library(stringr)#基因ID转换
library(enrichplot)#GO,KEGG,GSEA
library(clusterProfiler)#GO,KEGG,GSEA
library(GOplot)#弦图,弦表图,系统聚类图
library(DOSE)#
library(ggnewscale)#
library(topGO)#绘制通路网络图
library(circlize)#绘制富集分析圈图
library(ComplexHeatmap)#绘制图例
install.packages(&#34;/Users/ZYP/SCIENCE/SCRIPTS/R/orgdb/database/ZM4/org.ZZM4.eg.db&#34;, repos=NULL, type=&#34;sources&#34;)#安装自建库
library(org.ZZM4.eg.db)#读取自建库
################################Loading database################################
GO_database <- &#39;org.ZZM4.eg.db&#39;
KEGG_database <- &#39;/Users/ZYP/SCIENCE/SCRIPTS/R/orgdb/database/ZM4/KEGGdatabase_ZM4.RData&#39;
##################################Loading data##################################
info <- read.xlsx( &#34;/Users/ZYP/Downloads/diffexp_ZM4.xlsx&#34;, rowNames = F,colNames = T)#载入差异表达数据,只需ENTREZ ID和Log2FoldChange即可
genes <- info$Name
vector_genes <- as.vector(genes)
FCs <- info$Ratio
###################################GO富集分析###################################
GO<-enrichGO( genes,
OrgDb = GO_database,
keyType = &#34;GID&#34;,#设定读取的gene ID类型
ont = &#34;ALL&#34;,#(ont为ALL因此包括 Biological Process,Cellular Component,Mollecular Function三部分)
pvalueCutoff = 0.05,#设定p值阈值
minGSSize = 1,
# qvalueCutoff = 0.05,
# pAdjustMethod = &#39;BH&#39;
readable= FALSE)
##################################KEGG富集分析##################################
load(file = KEGG_database)
KEGG <- enricher(vector_genes,
TERM2GENE = pathway2gene,
TERM2NAME = pathway2name,
pvalueCutoff = 0.05,
# qvalueCutoff = 0.05,
# pAdjustMethod = &#34;BH&#34;,
minGSSize = 1)#基因集的最小基因数,最小最大默认是[10,500],在富集不显著时设置
#####################################可视化#####################################
barplot(GO, split=&#34;ONTOLOGY&#34;)+facet_grid(ONTOLOGY~., scale=&#34;free&#34;)#柱状图
barplot(KEGG,showCategory = 40,title = &#39;KEGG Pathway&#39;)
dotplot(GO, split=&#34;ONTOLOGY&#34;)+facet_grid(ONTOLOGY~., scale=&#34;free&#34;)#点状图
dotplot(KEGG)
enrichplot::cnetplot(GO,circular=FALSE,colorEdge = TRUE)#基因-通路关联网络图
enrichplot::cnetplot(KEGG,circular=FALSE,colorEdge = TRUE)#circluar为指定是否环化,基因过多时建议设置为FALSE
enrichplot::heatplot(GO,showCategory = 50)#基因-通路关联热图
enrichplot::heatplot(KEGG,showCategory = 50)
GO2 <- pairwise_termsim(GO)
KEGG2 <- pairwise_termsim(KEGG)
enrichplot::emapplot(GO2,showCategory = 50, color = &#34;p.adjust&#34;, layout = &#34;kk&#34;)#通路间关联网络图
enrichplot::emapplot(KEGG2,showCategory =50, color = &#34;p.adjust&#34;, layout = &#34;kk&#34;)
write.table(KEGG$ID, file = &#34;/Users/ZYP/Downloads/KEGG_IDs.txt&#34;, #将所有KEGG富集到的通路写入本地文件查看
append = FALSE, quote = TRUE, sep = &#34; &#34;,
eol = &#34;\n&#34;, na = &#34;NA&#34;, dec = &#34;.&#34;, row.names = TRUE,
col.names = TRUE, qmethod = c(&#34;escape&#34;, &#34;double&#34;),
fileEncoding = &#34;&#34;)
browseKEGG(KEGG,&#34;zmo02020&#34;)#选择其中的hsa05166通路进行展示
GO_BP<-enrichGO(genes,#GO富集分析BP模块
OrgDb = GO_database,
keyType = &#34;GID&#34;,
ont = &#34;BP&#34;,
pvalueCutoff = 0.05,
# pAdjustMethod = &#34;BH&#34;,
#qvalueCutoff = 0.05,
minGSSize = 1,
# maxGSSize = 500,
readable = T)
plotGOgraph(GO_BP)#GO-BP通路网络图
GO_CC<-enrichGO(genes,#GO富集分析CC模块
OrgDb = GO_database,
keyType = &#34;GID&#34;,
ont = &#34;CC&#34;,
pvalueCutoff = 0.05,
# pAdjustMethod = &#34;BH&#34;,
# qvalueCutoff = 0.05,
minGSSize = 1,
# maxGSSize = 500,
readable = T)
plotGOgraph(GO_CC)#GO-CC通路网络图
GO_MF<-enrichGO(genes,#GO富集分析MF模块
OrgDb = GO_database,
keyType = &#34;GID&#34;,
ont = &#34;MF&#34;,
pvalueCutoff = 0.05,
# pAdjustMethod = &#34;BH&#34;,
# qvalueCutoff = 0.05,
minGSSize = 1,
# maxGSSize = 500,
readable = T)
plotGOgraph(GO_MF)#GO-MF通路网络图
genedata<-data.frame(ID=genes,logFC=FCs)
write.table(GO$ONTOLOGY, file = &#34;/Users/ZYP/Downloads/GO_ONTOLOGYs.txt&#34;, #将所有GO富集到的基因集所对应的类型写入本地文件从而得到BP/CC/MF各自的起始位置如我的数据里是1,262,306
append = FALSE, quote = TRUE, sep = &#34; &#34;,
eol = &#34;\n&#34;, na = &#34;NA&#34;, dec = &#34;.&#34;, row.names = TRUE,
col.names = TRUE, qmethod = c(&#34;escape&#34;, &#34;double&#34;),
fileEncoding = &#34;&#34;)
GOplotIn_BP<-na.omit(GO[1:10,c(2,3,7,9)]) #提取GO富集BP的前10行,提取ID,Description,p,GeneID四列
GOplotIn_CC<-na.omit(GO[1:1,c(2,3,7,9)])#提取GO富集CC的前10行,提取ID,Description,p,GeneID四列
GOplotIn_MF<-na.omit(GO[41:50,c(2,3,7,9)])#提取GO富集MF的前10行,提取ID,Description,p,GeneID四列
GOplotIn_BP$geneID <-str_replace_all(GOplotIn_BP$geneID,&#39;/&#39;,&#39;,&#39;) #把GeneID列中的’/’替换成‘,’
GOplotIn_CC$geneID <-str_replace_all(GOplotIn_CC$geneID,&#39;/&#39;,&#39;,&#39;)
GOplotIn_MF$geneID <-str_replace_all(GOplotIn_MF$geneID,&#39;/&#39;,&#39;,&#39;)
names(GOplotIn_BP)<-c(&#39;ID&#39;,&#39;Term&#39;,&#39;adj_pval&#39;,&#39;Genes&#39;)#修改列名,后面弦图绘制的时候需要这样的格式
names(GOplotIn_CC)<-c(&#39;ID&#39;,&#39;Term&#39;,&#39;adj_pval&#39;,&#39;Genes&#39;)
names(GOplotIn_MF)<-c(&#39;ID&#39;,&#39;Term&#39;,&#39;adj_pval&#39;,&#39;Genes&#39;)
GOplotIn_BP$Category = &#34;BP&#34;#分类信息
GOplotIn_CC$Category = &#34;CC&#34;
GOplotIn_MF$Category = &#34;MF&#34;
circ_BP<-GOplot::circle_dat(GOplotIn_BP,genedata) #GOplot导入数据格式整理
circ_CC<-GOplot::circle_dat(GOplotIn_CC,genedata)
circ_MF<-GOplot::circle_dat(GOplotIn_MF,genedata)
chord_BP<-chord_dat(data = circ_BP,genes = genedata) #生成含有选定基因的数据框
chord_CC<-chord_dat(data = circ_CC,genes = genedata)
chord_MF<-chord_dat(data = circ_MF,genes = genedata)
GOChord(data = chord_BP,#弦图
title = &#39;GO-Biological Process&#39;,space = 0.01,#GO Term间距
limit = c(1,1),gene.order = &#39;logFC&#39;,gene.space = 0.25,gene.size = 5,
lfc.col = c(&#39;red&#39;,&#39;white&#39;,&#39;blue&#39;), #上下调基因颜色
process.label = 10) #GO Term字体大小
GOChord(data = chord_CC,title = &#39;GO-Cellular Component&#39;,space = 0.01,
limit = c(1,1),gene.order = &#39;logFC&#39;,gene.space = 0.25,gene.size = 5,
lfc.col = c(&#39;red&#39;,&#39;white&#39;,&#39;blue&#39;),
process.label = 10)
GOChord(data = chord_MF,title = &#39;GO-Mollecular Function&#39;,space = 0.01,
limit = c(1,1),gene.order = &#39;logFC&#39;,gene.space = 0.25,gene.size = 5,
lfc.col = c(&#39;red&#39;,&#39;white&#39;,&#39;blue&#39;),
process.label = 10)
GOCircle(circ_BP) #弦表图
GOCircle(circ_CC)
GOCircle(circ_MF)
chord<-chord_dat(data = circ_BP,genes = genedata) #生成含有选定基因的数据框
GOCluster(circ_BP,GOplotIn_BP$Term) #系统聚类图
chord<-chord_dat(data = circ_CC,genes = genedata)
GOCluster(circ_CC,GOplotIn_CC$Term)
chord<-chord_dat(data = circ_MF,genes = genedata)
GOCluster(circ_MF,GOplotIn_MF$Term)
########################
GSEA_input <- info$Ratio
names(GSEA_input) = info$Name
GSEA_input = sort(GSEA_input, decreasing = TRUE)
GSEA_KEGG <- gseKEGG(GSEA_input, organism = &#39;zmo&#39;, pvalueCutoff = 0.05)#GSEA富集分析
ridgeplot(GSEA_KEGG)
gseaplot2(GSEA_KEGG,1)
gseaplot2(GSEA_KEGG,1:30)#30是根据ridgeplot中有30个富集通路得到的
##################################富集分析圈图##################################
path <-&#39;/Users/ZYP/Downloads/&#39;
file <- &#39;GO&#39;
dat <- read.delim(str_c(path, file,&#39;.txt&#39;), sep = &#39;\t&#39;, row.names = 1, stringsAsFactors = FALSE, check.names = FALSE)
#首先给 ko id 排个序,默认按照原表格中的排列顺序
dat$id <- factor(rownames(dat), levels = rownames(dat))
##第一圈,绘制id
pdf(str_c(path,file,&#39;_circlize.pdf&#39;), width = 24, height = 12)
circle_size = unit(1, &#39;snpc&#39;)
circos.par(gap.degree = 0.5, start.degree = 90)
plot_data <- dat[c(&#39;id&#39;, &#39;gene_num.min&#39;, &#39;gene_num.max&#39;)] #选择作图数据集,定义了 ko 区块的基因总数量范围
#ko_color <- c(rep(&#39;#F7CC13&#39;,19), rep(&#39;#954572&#39;,21), rep(&#39;#0796E0&#39;,29)) #定义分组颜色
ko_color <- c( rep(&#39;#954572&#39;,1))
circos.genomicInitialize(plot_data, plotType = NULL, major.by = 1) #一个总布局
circos.track(
ylim = c(0, 1), track.height = 0.05, bg.border = NA, bg.col = ko_color, #圈图的高度、颜色等设置
panel.fun = function(x, y) {
ylim = get.cell.meta.data(&#39;ycenter&#39;) #ylim、xlim 用于指定 ko id 文字标签添加的合适坐标
xlim = get.cell.meta.data(&#39;xcenter&#39;)
sector.name = get.cell.meta.data(&#39;sector.index&#39;) #sector.name 用于提取 ko id 名称
circos.axis(h = &#39;top&#39;, labels.cex = 0.4, labels.niceFacing = FALSE) #绘制外周的刻度线
circos.text(xlim, ylim, sector.name, cex = 0.4, niceFacing = FALSE) #将 ko id 文字标签添加在图中指定位置处
} )
##第二圈,绘制富集的基因和富集 p 值
plot_data <- dat[c(&#39;id&#39;, &#39;gene_num.min&#39;, &#39;gene_num.rich&#39;, &#39;-log10Pvalue&#39;)] #选择作图数据集,包括富集基因数量以及 p 值等信息
label_data <- dat[&#39;gene_num.rich&#39;] #标签数据集,仅便于作图时添加相应的文字标识用
p_max <- round(max(dat$&#39;-log10Pvalue&#39;)) + 1 #定义一个 p 值的极值,以方便后续作图
colorsChoice <- colorRampPalette(c(&#39;#FF906F&#39;, &#39;#861D30&#39;)) #这两句用于定义 p 值的渐变颜色
color_assign <- colorRamp2(breaks = 0:p_max, col = colorsChoice(p_max + 1))
circos.genomicTrackPlotRegion(
plot_data, track.height = 0.08, bg.border = NA, stack = TRUE, #圈图的高度、颜色等设置
panel.fun = function(region, value, ...) {
circos.genomicRect(region, value, col = color_assign(value[[1]]), border = NA, ...) #区块的长度反映了富集基因的数量,颜色与 p 值有关
ylim = get.cell.meta.data(&#39;ycenter&#39;) #同上文,ylim、xlim、sector.name 等用于指定文字标签(富集基因数量)添加的合适坐标
xlim = label_data[get.cell.meta.data(&#39;sector.index&#39;),1] / 2
sector.name = label_data[get.cell.meta.data(&#39;sector.index&#39;),1]
circos.text(xlim, ylim, sector.name, cex = 0.4, niceFacing = FALSE) #将文字标签添(富集基因数量)加在图中指定位置处
} )
##第三圈,绘制上下调基因
#首先基于表格中上下调基因的数量,计算它们的占比
dat$all.regulated <- dat$up.regulated + dat$down.regulated
dat$up.proportion <- dat$up.regulated / dat$all.regulated
dat$down.proportion <- dat$down.regulated / dat$all.regulated
#随后,根据上下调基因的相对比例,分别计算它们在作图时的“区块坐标”和“长度”
dat$up <- dat$up.proportion * dat$gene_num.max
plot_data_up <- dat[c(&#39;id&#39;, &#39;gene_num.min&#39;, &#39;up&#39;)]
names(plot_data_up) <- c(&#39;id&#39;, &#39;start&#39;, &#39;end&#39;)
plot_data_up$type <- 1 #分配 1 指代上调基因
dat$down <- dat$down.proportion * dat$gene_num.max + dat$up
plot_data_down <- dat[c(&#39;id&#39;, &#39;up&#39;, &#39;down&#39;)]
names(plot_data_down) <- c(&#39;id&#39;, &#39;start&#39;, &#39;end&#39;)
plot_data_down$type <- 2 #分配 2 指代下调基因
#选择作图数据集(作图用)、标签数据集(添加相应的文字标识用),并分别为上下调基因赋值不同颜色
plot_data <- rbind(plot_data_up, plot_data_down)
label_data <- dat[c(&#39;up&#39;, &#39;down&#39;, &#39;up.regulated&#39;, &#39;down.regulated&#39;)]
color_assign <- colorRamp2(breaks = c(1, 2), col = c(&#39;red&#39;, &#39;blue&#39;))
#继续绘制圈图
circos.genomicTrackPlotRegion(
plot_data, track.height = 0.08, bg.border = NA, stack = TRUE, #圈图的高度、颜色等设置
panel.fun = function(region, value, ...) {
circos.genomicRect(region, value, col = color_assign(value[[1]]), border = NA, ...) #这里紫色代表上调基因,蓝色代表下调基因,区块的长度反映了上下调基因的相对占比
ylim = get.cell.meta.data(&#39;cell.bottom.radius&#39;) - 0.5 #同上文,ylim、xlim、sector.name 等用于指定文字标签(上调基因数量)添加的合适坐标
xlim = label_data[get.cell.meta.data(&#39;sector.index&#39;),1] / 2
sector.name = label_data[get.cell.meta.data(&#39;sector.index&#39;),3]
circos.text(xlim, ylim, sector.name, cex = 0.4, niceFacing = FALSE) #将文字标签(上调基因数量)添加在图中指定位置处
xlim = (label_data[get.cell.meta.data(&#39;sector.index&#39;),2]+label_data[get.cell.meta.data(&#39;sector.index&#39;),1]) / 2
sector.name = label_data[get.cell.meta.data(&#39;sector.index&#39;),4]
circos.text(xlim, ylim, sector.name, cex = 0.4, niceFacing = FALSE) #类似的操作,将下调基因数量的标签也添加在图中
} )
##第四圈,绘制富集因子
plot_data <- dat[c(&#39;id&#39;, &#39;gene_num.min&#39;, &#39;gene_num.max&#39;, &#39;rich.factor&#39;)] #选择作图数据集,包含富集因子列
label_data <- dat[&#39;category&#39;] #将通路的分类信息提取出,和下一句一起,便于作图时按分组分配颜色
color_assign <- c(&#39;BP&#39; = &#39;#F7CC13&#39;, &#39;CC&#39; = &#39;#954572&#39;, &#39;MF&#39; = &#39;#0796E0&#39;)
circos.genomicTrack(
plot_data, ylim = c(0, 1), track.height = 0.3, bg.col = &#39;gray95&#39;, bg.border = NA, #圈图的高度、颜色等设置
panel.fun = function(region, value, ...) {
sector.name = get.cell.meta.data(&#39;sector.index&#39;) #sector.name 用于提取 ko id 名称,并添加在下一句中匹配 ko 对应的高级分类,以分配颜色
circos.genomicRect(region, value, col = color_assign[label_data[sector.name,1]], border = NA, ytop.column = 1, ybottom = 0, ...) #绘制矩形区块,高度代表富集因子数值,颜色代表 ko 的分类
circos.lines(c(0, max(region)), c(0.5, 0.5), col = &#39;gray&#39;, lwd = 0.3) #可选在富集因子等于 0.5 的位置处添加一个灰线
} )
category_legend <- Legend(
labels = c(&#39;Cellular Component&#39;),
type = &#39;points&#39;, pch = NA, background = c(&#39;#954572&#39;),
#labels = c(&#39;Biological Process&#39;, &#39;Cellular Component&#39;, &#39;Molecular Function&#39;),
#type = &#39;points&#39;, pch = NA, background = c(&#39;#F7CC13&#39;, &#39;#954572&#39;, &#39;#0796E0&#39;),
labels_gp = gpar(fontsize = 8), grid_height = unit(0.5, &#39;cm&#39;), grid_width = unit(0.5, &#39;cm&#39;))
updown_legend <- Legend(
labels = c(&#39;Up-regulated&#39;, &#39;Down-regulated&#39;),
type = &#39;points&#39;, pch = NA, background = c(&#39;red&#39;, &#39;blue&#39;),
labels_gp = gpar(fontsize = 8), grid_height = unit(0.5, &#39;cm&#39;), grid_width = unit(0.5, &#39;cm&#39;))
pvalue_legend <- Legend(
col_fun = colorRamp2(round(seq(0, p_max, length.out = 6), 0),
colorRampPalette(c(&#39;#FF906F&#39;, &#39;#861D30&#39;))(6)),
legend_height = unit(3, &#39;cm&#39;), labels_gp = gpar(fontsize = 8),
title_gp = gpar(fontsize = 9), title_position = &#39;topleft&#39;, title = &#39;-Log10(Pvalue)&#39;)
lgd_list_vertical <- packLegend(category_legend, updown_legend, pvalue_legend)
grid.draw(lgd_list_vertical)
circos.clear()
dev.off()#保存
path <-&#39;/Users/ZYP/Downloads/ZM4/&#39;
file <- &#39;KEGG&#39;
dat <- read.delim(str_c(path, file,&#39;.txt&#39;), sep = &#39;\t&#39;, row.names = 1, stringsAsFactors = FALSE, check.names = FALSE)
#首先给 ko id 排个序,默认按照原表格中的排列顺序
dat$id <- factor(rownames(dat), levels = rownames(dat))
##第一圈,绘制id
pdf(str_c(path,file,&#39;_circlize.pdf&#39;), width = 24, height = 12)
circle_size = unit(1, &#39;snpc&#39;)
circos.par(gap.degree = 0.5, start.degree = 90)
plot_data <- dat[c(&#39;id&#39;, &#39;gene_num.min&#39;, &#39;gene_num.max&#39;)] #选择作图数据集,定义了 ko 区块的基因总数量范围
ko_color <- c(rep(&#39;#F7CC13&#39;,1), rep(&#39;#954572&#39;,1), rep(&#39;#0796E0&#39;,1)) #定义分组颜色
#ko_color <- c( rep(&#39;#954572&#39;,1))
circos.genomicInitialize(plot_data, plotType = NULL, major.by = 1) #一个总布局
circos.track(
ylim = c(0, 1), track.height = 0.05, bg.border = NA, bg.col = ko_color, #圈图的高度、颜色等设置
panel.fun = function(x, y) {
ylim = get.cell.meta.data(&#39;ycenter&#39;) #ylim、xlim 用于指定 ko id 文字标签添加的合适坐标
xlim = get.cell.meta.data(&#39;xcenter&#39;)
sector.name = get.cell.meta.data(&#39;sector.index&#39;) #sector.name 用于提取 ko id 名称
circos.axis(h = &#39;top&#39;, labels.cex = 0.4, labels.niceFacing = FALSE) #绘制外周的刻度线
circos.text(xlim, ylim, sector.name, cex = 0.4, niceFacing = FALSE) #将 ko id 文字标签添加在图中指定位置处
} )
##第二圈,绘制富集的基因和富集 p 值
plot_data <- dat[c(&#39;id&#39;, &#39;gene_num.min&#39;, &#39;gene_num.rich&#39;, &#39;-log10Pvalue&#39;)] #选择作图数据集,包括富集基因数量以及 p 值等信息
label_data <- dat[&#39;gene_num.rich&#39;] #标签数据集,仅便于作图时添加相应的文字标识用
p_max <- round(max(dat$&#39;-log10Pvalue&#39;)) + 1 #定义一个 p 值的极值,以方便后续作图
colorsChoice <- colorRampPalette(c(&#39;#FF906F&#39;, &#39;#861D30&#39;)) #这两句用于定义 p 值的渐变颜色
color_assign <- colorRamp2(breaks = 0:p_max, col = colorsChoice(p_max + 1))
circos.genomicTrackPlotRegion(
plot_data, track.height = 0.08, bg.border = NA, stack = TRUE, #圈图的高度、颜色等设置
panel.fun = function(region, value, ...) {
circos.genomicRect(region, value, col = color_assign(value[[1]]), border = NA, ...) #区块的长度反映了富集基因的数量,颜色与 p 值有关
ylim = get.cell.meta.data(&#39;ycenter&#39;) #同上文,ylim、xlim、sector.name 等用于指定文字标签(富集基因数量)添加的合适坐标
xlim = label_data[get.cell.meta.data(&#39;sector.index&#39;),1] / 2
sector.name = label_data[get.cell.meta.data(&#39;sector.index&#39;),1]
circos.text(xlim, ylim, sector.name, cex = 0.4, niceFacing = FALSE) #将文字标签添(富集基因数量)加在图中指定位置处
} )
##第三圈,绘制上下调基因
#首先基于表格中上下调基因的数量,计算它们的占比
dat$all.regulated <- dat$up.regulated + dat$down.regulated
dat$up.proportion <- dat$up.regulated / dat$all.regulated
dat$down.proportion <- dat$down.regulated / dat$all.regulated
#随后,根据上下调基因的相对比例,分别计算它们在作图时的“区块坐标”和“长度”
dat$up <- dat$up.proportion * dat$gene_num.max
plot_data_up <- dat[c(&#39;id&#39;, &#39;gene_num.min&#39;, &#39;up&#39;)]
names(plot_data_up) <- c(&#39;id&#39;, &#39;start&#39;, &#39;end&#39;)
plot_data_up$type <- 1 #分配 1 指代上调基因
dat$down <- dat$down.proportion * dat$gene_num.max + dat$up
plot_data_down <- dat[c(&#39;id&#39;, &#39;up&#39;, &#39;down&#39;)]
names(plot_data_down) <- c(&#39;id&#39;, &#39;start&#39;, &#39;end&#39;)
plot_data_down$type <- 2 #分配 2 指代下调基因
#选择作图数据集(作图用)、标签数据集(添加相应的文字标识用),并分别为上下调基因赋值不同颜色
plot_data <- rbind(plot_data_up, plot_data_down)
label_data <- dat[c(&#39;up&#39;, &#39;down&#39;, &#39;up.regulated&#39;, &#39;down.regulated&#39;)]
color_assign <- colorRamp2(breaks = c(1, 2), col = c(&#39;red&#39;, &#39;blue&#39;))
#继续绘制圈图
circos.genomicTrackPlotRegion(
plot_data, track.height = 0.08, bg.border = NA, stack = TRUE, #圈图的高度、颜色等设置
panel.fun = function(region, value, ...) {
circos.genomicRect(region, value, col = color_assign(value[[1]]), border = NA, ...) #这里紫色代表上调基因,蓝色代表下调基因,区块的长度反映了上下调基因的相对占比
ylim = get.cell.meta.data(&#39;cell.bottom.radius&#39;) - 0.5 #同上文,ylim、xlim、sector.name 等用于指定文字标签(上调基因数量)添加的合适坐标
xlim = label_data[get.cell.meta.data(&#39;sector.index&#39;),1] / 2
sector.name = label_data[get.cell.meta.data(&#39;sector.index&#39;),3]
circos.text(xlim, ylim, sector.name, cex = 0.4, niceFacing = FALSE) #将文字标签(上调基因数量)添加在图中指定位置处
xlim = (label_data[get.cell.meta.data(&#39;sector.index&#39;),2]+label_data[get.cell.meta.data(&#39;sector.index&#39;),1]) / 2
sector.name = label_data[get.cell.meta.data(&#39;sector.index&#39;),4]
circos.text(xlim, ylim, sector.name, cex = 0.4, niceFacing = FALSE) #类似的操作,将下调基因数量的标签也添加在图中
} )
##第四圈,绘制富集因子
plot_data <- dat[c(&#39;id&#39;, &#39;gene_num.min&#39;, &#39;gene_num.max&#39;, &#39;rich.factor&#39;)] #选择作图数据集,包含富集因子列
label_data <- dat[&#39;category&#39;] #将通路的分类信息提取出,和下一句一起,便于作图时按分组分配颜色
color_assign <- c(&#39;Sulfur metabolism&#39; = &#39;#F7CC13&#39;, &#39;elenocompound metabolism&#39; = &#39;#954572&#39;, &#39;Microbial metabolism in diverse environments&#39; = &#39;#0796E0&#39;)
circos.genomicTrack(
plot_data, ylim = c(0, 1), track.height = 0.3, bg.col = &#39;gray95&#39;, bg.border = NA, #圈图的高度、颜色等设置
panel.fun = function(region, value, ...) {
sector.name = get.cell.meta.data(&#39;sector.index&#39;) #sector.name 用于提取 ko id 名称,并添加在下一句中匹配 ko 对应的高级分类,以分配颜色
circos.genomicRect(region, value, col = color_assign[label_data[sector.name,1]], border = NA, ytop.column = 1, ybottom = 0, ...) #绘制矩形区块,高度代表富集因子数值,颜色代表 ko 的分类
circos.lines(c(0, max(region)), c(0.5, 0.5), col = &#39;gray&#39;, lwd = 0.3) #可选在富集因子等于 0.5 的位置处添加一个灰线
} )
category_legend <- Legend(
#labels = c(&#39;Cellular Component&#39;),
#type = &#39;points&#39;, pch = NA, background = c(&#39;#954572&#39;),
labels = c(&#39;Sulfur metabolism&#39;, &#39;elenocompound metabolism&#39;, &#39;Microbial metabolism in diverse environ&#39;),
type = &#39;points&#39;, pch = NA, background = c(&#39;#F7CC13&#39;, &#39;#954572&#39;, &#39;#0796E0&#39;),
labels_gp = gpar(fontsize = 8), grid_height = unit(0.5, &#39;cm&#39;), grid_width = unit(0.5, &#39;cm&#39;))
updown_legend <- Legend(
labels = c(&#39;Up-regulated&#39;, &#39;Down-regulated&#39;),
type = &#39;points&#39;, pch = NA, background = c(&#39;red&#39;, &#39;blue&#39;),
labels_gp = gpar(fontsize = 8), grid_height = unit(0.5, &#39;cm&#39;), grid_width = unit(0.5, &#39;cm&#39;))
pvalue_legend <- Legend(
col_fun = colorRamp2(round(seq(0, p_max, length.out = 6), 0),
colorRampPalette(c(&#39;#FF906F&#39;, &#39;#861D30&#39;))(6)),
legend_height = unit(3, &#39;cm&#39;), labels_gp = gpar(fontsize = 8),
title_gp = gpar(fontsize = 9), title_position = &#39;topleft&#39;, title = &#39;-Log10(Pvalue)&#39;)
lgd_list_vertical <- packLegend(category_legend, updown_legend, pvalue_legend)
grid.draw(lgd_list_vertical)
circos.clear()
dev.off()#保存





<hr/>欢迎私信学术交流与合作。 |
|



 发表于 2023-1-17 04:01:34
发表于 2023-1-17 04:01:34